期刊文章
过刊浏览
- Volumes 84-95 (2024)
-
Volumes 72-83 (2023)
-
Volume 83
Pages 1-258 (December 2023)
-
Volume 82
Pages 1-204 (November 2023)
-
Volume 81
Pages 1-188 (October 2023)
-
Volume 80
Pages 1-202 (September 2023)
-
Volume 79
Pages 1-172 (August 2023)
-
Volume 78
Pages 1-146 (July 2023)
-
Volume 77
Pages 1-152 (June 2023)
-
Volume 76
Pages 1-176 (May 2023)
-
Volume 75
Pages 1-228 (April 2023)
-
Volume 74
Pages 1-200 (March 2023)
-
Volume 73
Pages 1-138 (February 2023)
-
Volume 72
Pages 1-144 (January 2023)
-
Volume 83
-
Volumes 60-71 (2022)
-
Volume 71
Pages 1-108 (December 2022)
-
Volume 70
Pages 1-106 (November 2022)
-
Volume 69
Pages 1-122 (October 2022)
-
Volume 68
Pages 1-124 (September 2022)
-
Volume 67
Pages 1-102 (August 2022)
-
Volume 66
Pages 1-112 (July 2022)
-
Volume 65
Pages 1-138 (June 2022)
-
Volume 64
Pages 1-186 (May 2022)
-
Volume 63
Pages 1-124 (April 2022)
-
Volume 62
Pages 1-104 (March 2022)
-
Volume 61
Pages 1-120 (February 2022)
-
Volume 60
Pages 1-124 (January 2022)
-
Volume 71
- Volumes 54-59 (2021)
- Volumes 48-53 (2020)
- Volumes 42-47 (2019)
- Volumes 36-41 (2018)
- Volumes 30-35 (2017)
- Volumes 24-29 (2016)
- Volumes 18-23 (2015)
- Volumes 12-17 (2014)
- Volume 11 (2013)
- Volume 10 (2012)
- Volume 9 (2011)
- Volume 8 (2010)
- Volume 7 (2009)
- Volume 6 (2008)
- Volume 5 (2007)
- Volume 4 (2006)
- Volume 3 (2005)
- Volume 2 (2004)
- Volume 1 (2003)
Volume 10 Issue 3
Zhang, Q., Ren, H., Wang, W., Zhang, J., & Zhang, H. (2012). Molecular simulation of oligomer inhibitors for calcite scale. Particuology, 10(3), 266–275. https://doi.org/10.1016/j.partic.2011.04.004
Molecular simulation of oligomer inhibitors for calcite scale
Qiuyu Zhang *, Hua Ren, Wenwen Wang, Junping Zhang, Hepeng Zhang
School of Science, Northwestern Polytechnical University, Xi’an 710072, China
10.1016/j.partic.2011.04.004
Volume 10, Issue 3,
June 2012,
Pages 266-275
Received 24 October 2010, Revised 11 March 2011, Accepted 21 April 2011, Available online 13 July 2011.
E-mail:
qyzhang1803@gmail.com
Highlights
Abstract
Molecular simulation was performed to study the interaction between CaCO3 crystal and several oligomer inhibitors, by using the equilibrium morphology method to calculate the growth morphology of CaCO3 without inhibitors. The calculated morphology agreed well with SEM photographs. Then, a double-layer model was built to investigate the interaction between calcite crystal and oligomer inhibitors containing maleic anhydride (MA) and acrylic acid (AA). Interaction energy per gram of an oligomer inhibitor was introduced as a scale of inhibition efficiency of different monomers. The results indicated that, for calcite scale inhibition, acrylamide (AM) and vinyl phosphonic acid (VPA) were the most efficient monomers, while allylsulfonic acid (AS) was the poorest. Increasing proportion of AM in dimer inhibitor molecule would improve the inhibition efficiency of MA, though, for a trimer, such as MA–AA–AM, certain sequence of monomers in the inhibitor molecule was necessary besides higher proportion of AM.
Graphical abstract
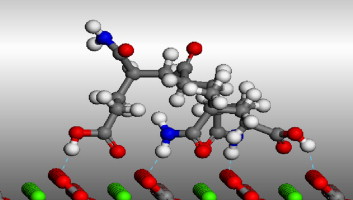
Keywords
Molecular simulation; Crystal morphology; Interaction energy; Scale inhibitor; Scale inhibition efficiency
文章导读